
Big Data, une approche pour mesurer la fiabilité des arbres phylogénétiques
Une publication dans Nature de l’Institut Pasteur et du CNRS, en association avec des chercheurs d’Afrique du Sud, propose une nouvelle méthode de bootstrap phylogénétique. Cet article fait suite à l’une des publications les plus citées de l’histoire des sciences (> 35 000 citations), rédigée par Joseph Felsenstein en 1985. Cette publication décrivait la première méthode de bootstrap phylogénétique, qui depuis plus de 30 ans s’est avérée extrêmement utile et pertinente dans de très nombreux domaines. Cependant, avec l’arrivée des données biologiques massives et le séquençage à haut débit, la méthode de Felsenstein montre des limites sérieuses et s’avère souvent incapable de révéler le signal contenu dans les grands jeux de données. La méthode proposée par l’équipe d’Olivier Gascuel, du Centre de Bioinformatique, Biostatistique et Biologie Intégrative, s’attaque précisément à cette limitation. L’article de Nature démontre ses performances sur de grands alignements de séquences issues des mammifères et du VIH-SIDA, ainsi que sur des simulations.
Theodosius Dobzhansky fût, dans les années 30, l’un des premiers à comprendre les liens entre les théories de Darwin et la génétique. En 1973 il publiait un essai fameux intitulé « Nothing in Biology Makes Sense Except in the Light of Evolution » (rien en biologie n’a de sens, excepté à la lumière de l’évolution). Avec l’irruption des données massives issues du séquençage de l’ADN, ses prédictions se confirment : les méthodes évolutives s’avèrent indispensables pour étudier et comprendre de nombreux objets biologiques, depuis le niveau moléculaire, la génomique fonctionnelle et les familles de protéines, jusqu’au niveau populationnel et aux écosystèmes, en passant, par exemple, par la compréhension et le suivi des épidémies. Un outil essentiel dans ces études est la reconstruction phylogénétique : en utilisant des séquences d’ADN ou protéiques, on reconstruit des arbres évolutifs au sens de Darwin, qui représentent les filiations et la généalogie des objets biologiques étudiés —on parle dans le domaine de taxons pour « taxonomic units ». On utilise des séquences dites « homologues » qui descendent d’une même séquence ancestrale. Les variations de ces séquences permettent de reconstruire leur histoire et leurs proximités évolutives et par là celles des taxons auxquels elles appartiennent. Les méthodes de reconstruction phylogénétique ont beaucoup progressé depuis une trentaine d’année, et on dispose aujourd’hui d’algorithmes très performants pour inférer des arbres phylogénétiques à partir de séquences. Les algorithmes et logiciels phylogénétiques font partie des incontournables de la bioinformatique, notamment PhyML (Guindon et Gascuel 2003 ; Guindon et al. 2010 ; LIRMM UMR 5506) qui a été cité environ 20 000 fois (cf. Google Scholar).
Une fois les phylogénies reconstruites, se pose la question de leur fiabilité. Quelles parties de l’arbre peuvent être considérées comme sûres, et quelles autres ne reflètent que le bruit inhérent aux données ? La situation est analogue à celle qu’on rencontre avec les estimations numériques, pour lesquelles on calcule des barres d’erreur et des intervalles de confiance. Mais le problème est plus ardu car l’estimation est un arbre, un objet mathématique autrement plus complexe qu’une simple valeur numérique. Joseph Felsenstein a été le premier à proposer une méthode pertinente, inspirée du bootstrap statistique usuel, qui avait été introduit et développé par Bradley Efron au début des années 80. On génère une variabilité en ré-échantillonnant les données originelles et ces pseudo-échantillons ou « échantillons bootstrap » sont ensuite utilisés pour calculer de nouvelles estimations, produisant ainsi une distribution des valeurs estimées, plutôt qu’une unique estimation ponctuelle correspondant à l’échantillon d’origine. Le mécanisme de ré-échantillonnage le plus commun consiste à tirer avec remise les observations de l’échantillon originel, pour simuler ainsi le tirage d’échantillons dans une population plus vaste. La méthode proposée par Felsenstein est similaire : les pseudo-échantillons (issus du jeu initial de séquences) sont utilisés pour inférer des pseudo-arbres ou « arbres bootstrap », qui sont comparés à l’arbre originel. Les branches de l’arbre originel retrouvées dans une forte proportion d’arbres bootstrap ont un fort support statistique, et inversement les branches peu ou pas retrouvées ont un support faible. La pertinence, la simplicité et l’interprétabilité de cette méthode l’ont rendu extrêmement courante dans les études évolutives, au point qu’elle est généralement requise pour la publication de phylogénies. Avec 35 000 citations, l’article de Felsenstein est classé dans le top 100 des articles scientifiques les plus cités de tous les temps. En 2017, il a été cité plus de 2 000 fois (cf. Google Scholar).
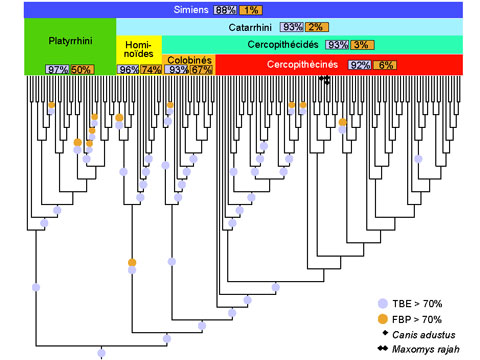
Cependant, les supports issus du bootstrap de Felsenstein (FBP, pour Felsenstein Bootstrap Proportion) ont tendance à être très faibles dès lors que les arbres phylogénétiques portent sur un nombre élevé de taxons, tandis que les grands jeux de données contenant des centaines ou des milliers de séquences sont maintenant communs grâce aux technologies de séquençage à haut débit. La raison de cette dégradation est expliquée par la méthodologie même du bootstrap de Felsenstein. Une branche d’un arbre bootstrap doit correspondre exactement à une branche de l’arbre d’origine, pour être prise en compte. Une branche d’un arbre partitionne en deux les taxons étudiés, suivant qu’ils sont situés d’un côté ou l’autre de la branche ; deux branches sont identiques si elles induisent la même partition. Dans la méthode de Felsenstein, une différence d’un seul taxon suffit pour que la branche bootstrap soit comptée absente, alors qu’elle est presque identique à la branche d’origine. L’approche standard consiste à supprimer les taxons phylogénétiquement instables, et à relancer l’analyse. Mais cela est statistiquement discutable et coûteux en termes de calculs. De plus, avec les grands arbres toutes les branches de l’arbre sont susceptibles d’être (légèrement) erronées, et une grande partie des taxons peut être relativement instable.
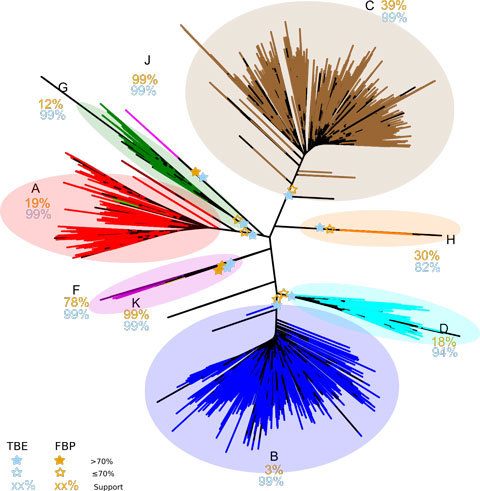
L’article de Nature propose une nouvelle version du bootstrap phylogénétique, dans laquelle la présence des branches originelles dans les arbres bootstrap est mesurée grâce à une distance de « transfert » graduelle, contrairement à la version de Felsenstein qui utilise un index binaire de présence / absence. Cette distance de transfert est normalisée puis moyennée sur tous les arbres bootstrap. On obtient ainsi le support de branche « TBE » (pour Transfert Bootstrap Expectation), par construction plus élevé que les supports FBP. Lorsqu’il est combiné avec une méthode d’inférence statistiquement consistante, TBE supporte très rarement des branches fortement erronées. Les résultats avec des données de séquences issues des mammifères et du VIH-SIDA, ainsi que des données simulées, démontrent clairement l’utilité de l’approche, en particulier concernant les branches profondes et les grands arbres, où des branches connues pour être essentiellement correctes sont supportées par TBE mais pas par FBP. Les supports TBE sont aisément interprétables comme des fractions de taxons instables, et la capacité de TBE à identifier les taxons les plus instables (par exemple les séquences recombinantes du VIH) permet d’étudier ces taxons plus précisément, de comprendre pourquoi ils sont phylogénétiquement instables et de réviser les supports de branche et la phylogénie dans son ensemble. La méthode est implémentée dans un serveur web et le code C est disponible en open source (http://booster.c3bi.pasteur.fr/).
Référence :
F. Lemoine, J.-B. Domelevo Entfellner, E. Wilkinson, D. Correia, M. Dávila Felipe, T. De Oliveira, O. Gascuel. Renewing Felsenstein’s Phylogenetic Bootstrap in the Era of Big Data, Nature to be completed, 2018.