
Un dixième gène pour le virus du SIDA
Selon les manuels de biologie, le virus du SIDA (le VIH) a neuf gènes. Suggérée à la fin des années 1980, l’existence d’un dixième gène, asp, restait débattue ; elle vient d’être confirmée par des chercheurs qui ont comparé 23 000 séquences du VIH et du SIV (l’équivalent du VIH chez les singes). Ils montrent que le gène asp n’existe que chez le groupe de virus responsable de la pandémie humaine et que son apparition est concomitante à l’émergence du VIH-1 chez l’homme, vers le début du XXe siècle. Ces travaux, publiés cette semaine dans PNAS, mettent aussi en évidence une forte pression de sélection sur le gène asp. Autrement dit, son maintien au sein du génome viral est favorisé, ce qui signifie qu’il confère un avantage au virus. Il reste maintenant à trouver quelle est sa fonction exacte.
Les chercheurs ont réussi cette prouesse grâce à une nouvelle approche bioinformatique. En effet, l’étude de ce gène est compliquée par le fait que sa séquence (chevauchante) est codée sur le brin antisens du génome proviral du VIH-1, et entièrement incluse dans la région du gène env codant pour l’enveloppe du virus.
Le VIH est un retrovirus. Son matériel génétique est codé dans un ARN qui est rétrotranscrit en ADN puis intégré dans l’ADN génomique de l’hôte humain. Ce « génome proviral » a deux brins : le brin sens correspondant à l’ARN du VIH, et le brin antisens, l’ensemble formant la double hélice de l’ADN. On sait depuis plusieurs années que les rétrovirus sont capables de transcrire les deux brins de leur ADN proviral. La difficulté concernant ce 10e gène du VIH, nommé asp tout comme la protéine dont il permet la production (ASP signifiant AntiSense Protein), venait de ce que ce gène est en chevauchement du gène env qui code pour l’enveloppe virale. En effet, le code génétique est redondant : dans les gènes usuels (non-chevauchants), chaque codon (séquence de trois nucléotides) code pour un unique acide aminé, et la plupart des acides aminés peuvent être codés par plusieurs codons différents. La redondance du code génétique est à la base des approches classiques pour étudier la pression de sélection. Avec les gènes chevauchants au contraire, une même portion de séquence code pour deux (voire trois) protéines. Comme si un mot français avait plusieurs sens possibles lorsqu’on décale le début de la lecture, ou qu’on le lit à l’envers. Le codage de l’information génétique est alors sur-contraint : les deux protéines chevauchantes sont fortement corrélées et de nombreuses combinaisons sont impossibles. De ce fait, les approches bioinformatiques standards étaient largement inopérantes.
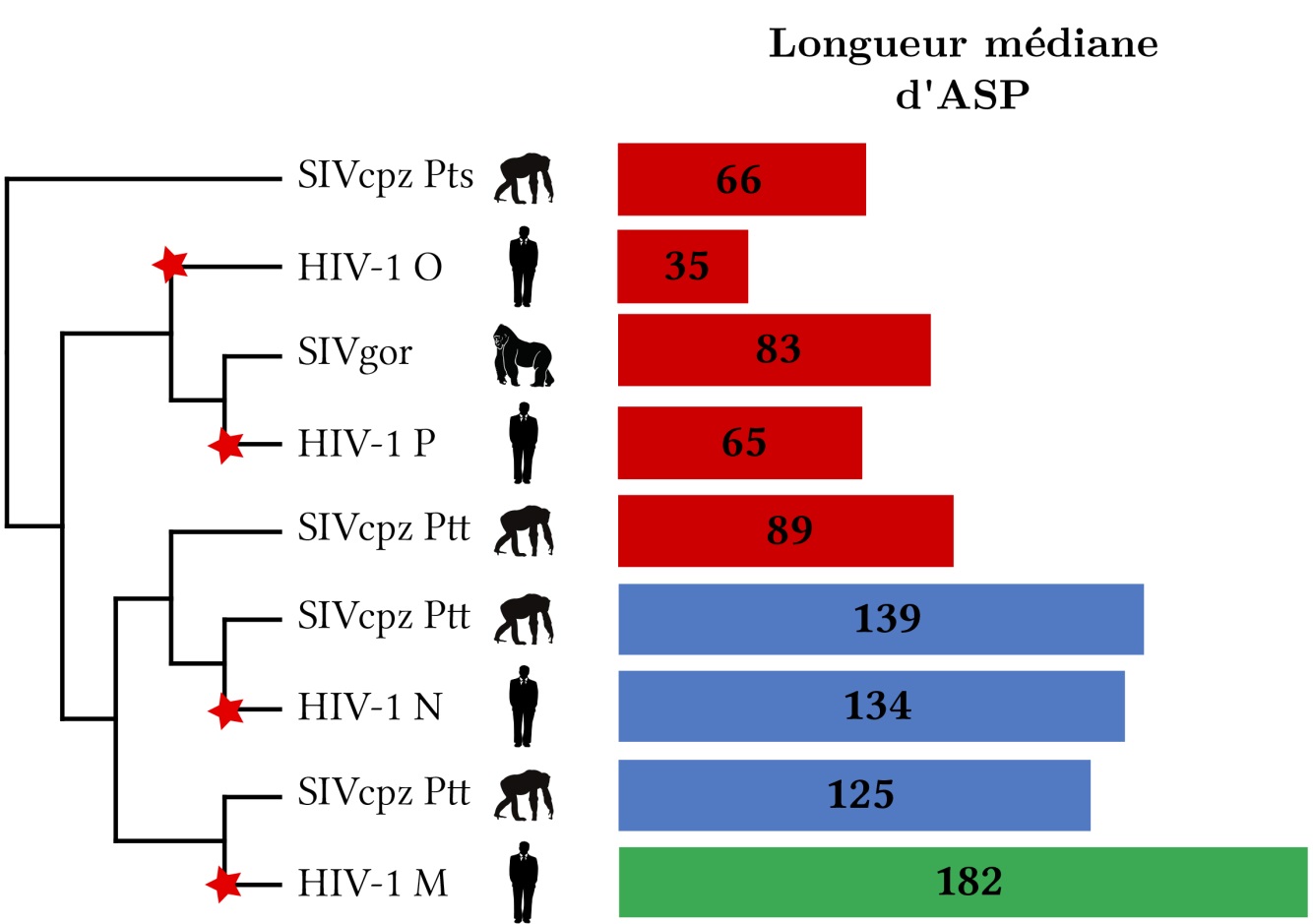
Dans le cadre d’un projet (PEPS de site) soutenu par l’Université de Montpellier et la Mission pour l’interdisciplinarité du CNRS, des chercheurs du LIRMM, spécialisés dans les modèles mathématiques de l’évolution biologique, et du CPBS, experts dans la transcription antisens chez les rétrovirus, ont réalisé la première étude évolutive du gène asp. L’objectif était d’analyser la conservation et l’évolution d’asp, depuis l’origine de l’introduction du VIH-1 chez l’homme. Les chercheurs ont donc étudié près de 23 000 séquences du virus afin de déterminer si le gène asp était conservé dans les différents groupes du VIH-1 et du SIV, et de mesurer s’il existait une pression de sélection agissant sur asp. Pour cela, une nouvelle méthode originale a été développée, basée sur l’étude fine des codons stops. Les codons stopscorrespondent à des signaux d’arrêt dans le mécanisme de synthèse des protéines et ne sont donc pas observés à l’intérieur des gènes. Les chercheurs ont montré que dans le cas du gène asp, certains stopsétaient « obligatoires » et imposés par la protéine chevauchante Env, alors que d’autres étaient « potentiels » et pouvaient apparaître ou disparaître sans que Env soit modifiée. En l’absence de pression de sélection, on s’attendait à ce que la présence effective de ces stops potentiels soit aléatoire dans la région d’asp. A l’inverse, si le gène et la protéine ASP étaient fonctionnels et utiles au virus, on s’attendait à ce que les stops potentiels soient systématiquement évités. En utilisant l’ensemble des 23 000 séquences du VIH-1 publiquement disponibles chez le gorille, le chimpanzé et l’homme, les chercheurs ont montré que les stops potentiels étaient effectivement évités dans le groupe M du VIH-1 à l’origine de la pandémie humaine. Cela indiquait une forte pression de sélection, alors que ces codons stops étaient présents aléatoirement dans les autres groupes du VIH-1 ainsi que chez les SIV des singes. D’autres résultats sont venus conforter cette analyse. En utilisant des simulations informatiques, les chercheurs ont montré que le taux de présence de asp au sein du groupe pandémique M avait une probabilité quasi-nulle d’être obtenu par hasard. Ils ont également observé que dans certains sous-types viraux une mutation ayant fait disparaître asp avait été compensée par une nouvelle mutation pour continuer à synthétiser la protéine ASP.
La particularité du positionnement de ce gène, en antisens et en recouvrement du gène de l’enveloppe virale, ne permettait pas jusque-là de caractériser une pression évolutive avec les méthodes de bioinformatique conventionnelles. C’est donc grâce aux avancées en bioinformatique évolutive que la pression de sélection en faveur du maintien du gène asp dans le groupe M a pu être mise en évidence. La fonction d’ASP dans la multiplication virale ainsi que son rôle exact dans la pandémie de SIDA restent pour autant largement inconnus. A la suite de cette étude de nouvelles recherches devraient émerger, visant à comprendre le rôle d’ASP dans l’infection par le VIH-1. Les méthodes bioinformatiques développées permettront aussi l’analyse systématique d’autre virus chez lesquels des gènes chevauchants seraient passés inaperçus ou nécessiteraient des confirmations évolutives.
Les laboratoires à l’origine de cette découverte
L’équipe du LIRMM est à l’origine de plusieurs logiciels de phylogénie, c’est-à-dire l’étude des relations de parenté entre objets biologiques (espèces, organelles, gènes, protéines…), qui sont très diffusés, notamment PhyML qui recueille plus de 15 000 citations. Leurs travaux portent sur la biologie évolutive en général, la modélisation dans ce domaine, et depuis une dizaine d’années les pathogènes humains.
L’équipe du CPBS quant à elle a démontré il y a une quinzaine d’années que chez le rétrovirus HTLV-1, un virus proche du VIH-1, le brin antisens permettait la synthèse de la protéine HBZ, essentielle à l’établissement de la chronicité et responsable du développement d’une leucémie T chez l’adulte infecté. L’équipe du CPBS est engagée depuis plusieurs années dans les recherches sur asp. Elle a démontré l’existence de transcrits et étudié leur régulation.
Publication :
Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic, Élodie Cassan1
2
, Anne-Muriel Arigon-Chifolleau1
, Jean-Michel Mesnard2
, Antoine Gross2
et Olivier Gascuel1
3
.
- 1 a b c Équipe Méthodes et Algorithmes pour la Bioinformatique du Laboratoire d’Informatique, de Robotique et de Microélectronique de Montpellier (LIRMM - CNRS/Université de Montpellier)
- 2 a b c Équipe Acteur de la Pathogénèse des Infections Rétrovirales du Centre d’études d’agents Pathogènes et Biotechnologies pour la Santé (CPBS - CNRS/Université de Montpellier)
- 3 Unité Bioinformatique Évolutive du Centre de Bioinformatique, Biostatistique et Biologie Intégrative (C3BI – CNRS/Institut Pasteur)