PhyML, un logiciel pour remonter la piste du coronavirus
Face à un virus encore mal connu, la recherche a besoin de tous les moyens pour lutter contre le COVID-19. Conçu, hébergé et maintenu à jour par des chercheurs et des ingénieurs du LIRMM (CNRS/Université de Montpellier), PhyML est un logiciel qui compare les séquences d’ADN pour en tirer des arbres phylogénétiques et expliquer l’évolution qui a permis leur apparition. Partout dans le monde, des équipes s’en emparent pour remonter le fil de l’épidémie.
Utilisé par les biologistes depuis plus de quinze ans, le logiciel PhyML fait partie de l’arsenal d’outils disponibles pour mieux comprendre et combattre le coronavirus SARS-CoV-2, responsable de l’épidémie COVID-19. Créé et maintenu par Stéphane Guindon, chargé de recherche CNRS au Laboratoire d’informatique, de robotique et de microélectronique de Montpellier (LIRMM, CNRS/Université de Montpellier), PhyML compare des séquences génétiques pour établir leurs liens de parentés évolutives.
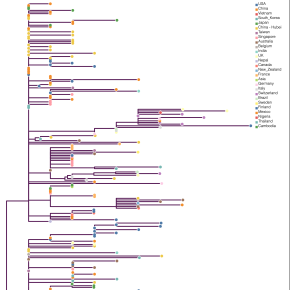
Début février, PhyML a été appliqué à la comparaison de cinquante-six génomes complets de souches de SARS-CoV-2, soit autant de chaînes d’environ 30 000 nucléotides. Ces travaux ont montré que l’origine de l’épidémie provenait d’un premier groupe d’infectés à Wuhan, début décembre 2019, à la suite d’une seule contamination par un animal. L’analyse phylogénétique confirme qu’il n’y a pas eu ensuite d’autres transmissions de l’animal à l’humain. Le corpus a depuis été porté à 176 souches virales, et continue de grandir. Ces études sont menées par différentes équipes internationales, par exemple les universités d’Édimbourg ou de Melbourne.
PhyML a cependant besoin de signal pour fonctionner : lorsque les séquences sont trop similaires, l’arbre phylogénétique ne peut pas être reconstruit avec précision. C’est d’ailleurs une des difficultés rencontrées dans l’analyse du SARS-Cov-2. Le virus étant extrêmement récent, il a peu muté et présente donc une diversité génétique encore relativement faible. Il livre moins facilement ses secrets : par exemple, certaines souches françaises et chinoises sont à peine discernables.
Cela n’empêche bien sûr pas la recherche d’avancer. Une publication dans la revue Nature a ainsi réfuté les théories selon lesquelles SARS-CoV-2 aurait été fabriqué en laboratoire. Les scientifiques ont repéré les mutations qui ont permis au virus d’attaquer aussi efficacement les humains. Reste encore à savoir si cette mutation est apparue avant ou après la transmission à notre espèce, car le premier cas multiplierait les risques de réémergences de la maladie.
Créé en 2003 à partir des travaux de thèse de Stéphane Guindon, encadrés par Olivier Gascuel
Le logiciel est presque quotidiennement mis à jour grâce aux retours d’une large communauté d’utilisateurs. Il est en effet cité dans pas moins de 25 000 publications scientifiques, principalement liées à la biologie et à l’étude de l’évolution. Les améliorations visent surtout à implémenter des algorithmes de calcul plus rapides, et à s’adapter à l’accroissement du volume des données.
Pour utiliser PhyML, les chercheurs du monde entier déposent leurs séquences sur le site dédié
La classification phylogénétique servait à l’origine uniquement à classifier les espèces, mais elle va maintenant bien plus loin. Avec l’amélioration du traitement des données moléculaires, les approches phylogénétiques fournissent aussi des estimations des taux auxquels les espèces vivantes apparaissent et s’éteignent, ou la taille de populations au sein d’une famille d’espèces. Elles permettent par ailleurs de déterminer l’origine géographique des évènements de contamination. « Les arbres phylogénétiques nous donnent la possibilité de remonter les chaînes de transmission virale », précise Stéphane Guindon.
Avec ses collègues, il aimerait à présent intégrer PhyML à un « tableau de bord » de suivi d’épidémies. Les chercheurs veulent notamment visualiser les arbres phylogénétiques de manière dynamique et les combiner à différentes informations géographiques, ainsi que d’autres données disponibles à propos l’épidémie. Cet outil intégré faciliterait le suivi de l’épidémie à grande échelle et aiderait les épidémiologistes à mieux comprendre sa dynamique temporelle et spatiale.